Progeria, l'un des nombreux troubles humains rares associés au vieillissement prématuré. Les deux principaux types de progeria sont le syndrome de progeria de Hutchinson-Gilford (HGPS), qui a son apparition dans la petite enfance, et le syndrome de Werner (progeria d'adulte), qui survient plus tard dans la vie. Une troisième condition, le syndrome de Hallerman-Streiff-François, est caractérisée par la présence de progeria en combinaison avec nanisme et autres caractéristiques de croissance anormale. La progeria est extrêmement rare; par exemple, l'incidence mondiale du syndrome de progeria Hutchinson-Gilford est d'environ un sur quatre à huit millions de naissances.
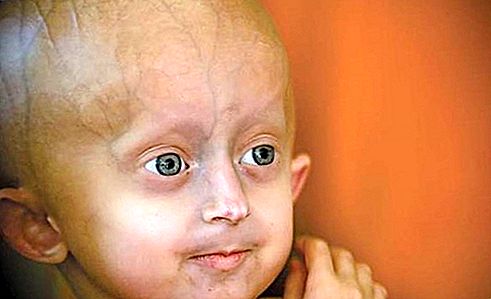
Les signes du syndrome de progeria Hutchinson-Gilford apparaissent vers l'âge d'un an, après une enfance manifestement normale. Les personnes affectées dépassent rarement la taille d'un enfant de 5 ans normal, bien qu'elles aient l'apparence physique d'adultes de 60 ans au moment où elles ont 10 ans. Bon nombre des aspects superficiels du vieillissement, tels que la calvitie, l'amincissement de la la peau, la proéminence des vaisseaux sanguins du cuir chevelu et les maladies vasculaires se produisent. Les organes sexuels restent petits et sous-développés. Quelques individus atteints de progeria sont intellectuellement handicapés, mais la plupart ont une intelligence normale et peuvent même être précoces. À l'âge de 10 ans, une artériosclérose étendue et des maladies cardiaques se sont développées, et la plupart des patients meurent avant l'âge de 30 ans; l'âge médian de décès est de 13 ans. La condition n'est pas héréditaire. Il résulte plutôt de la mutation spontanée d'un gène appelé LMNA (lamin A / C). Une seule copie du gène muté est nécessaire dans chaque cellule pour provoquer la maladie; il s'agit donc d'un trouble autosomique dominant.

Le syndrome de Werner apparaît généralement après la puberté, les signes visibles de vieillissement devenant plus apparents après l'âge de 20 ans. Les individus peuvent ne pas atteindre leur taille adulte projetée, car la poussée de croissance de l'adolescence peut être émoussée. Les patients atteints du syndrome de Werner sont sexuellement matures, mais les caractéristiques sexuelles secondaires sont peu développées. Les signes superficiels du vieillissement sont la calvitie ou le grisonnement prématuré des cheveux, la perte des dents et de l'ouïe, les cataractes, les épisodes arthritiques aigus, les ulcères cutanés et l'ostéoporose (perte de tissu osseux). Il y a une tendance accrue à développer des maladies cardiaques, le diabète sucré et le cancer, et la durée de vie moyenne est de 47 ans. Le syndrome de Werner est provoqué par des mutations du gène WRN (syndrome de Werner, semblable à l'hélicase RecQ). Ce type de progeria est héréditaire et se transmet comme un trait récessif (deux copies du gène muté, une de chaque parent, sont nécessaires pour provoquer la maladie).
Initialement, la progeria a été étudiée comme un modèle de vieillissement normal, mais, comme tous les organes ne sont pas affectés, cela n'est plus considéré comme approprié. Il n'y a, par exemple, aucun signe de sénilité ou de vieillissement dans le système nerveux central. Il existe des preuves expérimentales suggérant que le traitement avec un médicament connu sous le nom de n-acétylcystéine pourrait ralentir le processus de vieillissement associé au syndrome de progeria Hutchinson-Gilford.












